
图文摘要
成果简介
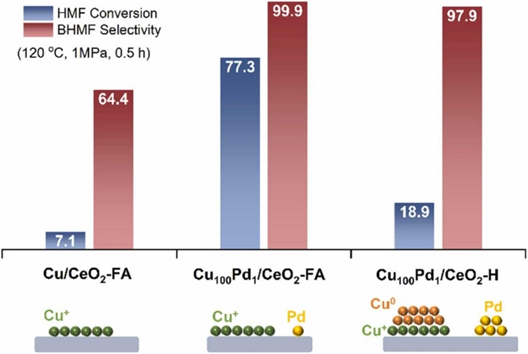
近日,中国科学院宁波材料技术与工程研究所张建课题组在Applied Catalysis B: Environment and Energy(影响因子20.2)上发表了题为“Boosting hydrogenation properties of supported Cu-based catalysts by replacing Cu0 active sites”的研究论文(DOI:10.1016/j.apcatb.2024.124563)。平衡Cu+/Cu0比率是提高铜基催化剂催化活性的常见策略,但仍然受到低原子利用率和电荷分布固有性质的限制,该文提出在二氧化铈上构建双金属位点来替代铜基催化剂中的Cu0活性位点的策略,该双金属位点由空间上分离的痕量钯金属和具有单原子层的板状Cu+团簇组成。制备的Cu100Pd1/CeO2-FA催化剂(使用甲酸)对5-羟甲基糠醛选择性加氢制2,5-双(羟甲基)呋喃的催化活性是传统Cu100Pd1/CeO2-H催化剂的4倍,甚至优于现有的一些贵金属催化剂。
全文速览
本研究成功合成了一种双金属催化剂,其特征是空间分离的痕量钯金属和CeO2上具有单原子层的片状Cu+团簇。其中,Cu100Pd1/CeO2-FA(Pd/Cu=1:100,mol/mol)对HMF选择性加氢具有显著的催化活性。多重表征和理论计算表明,Pd原子是H2分子的异解活化位点,而板状Cu+金属簇是有效的加氢位点。这种涉及活性位点的空间关系和电子结构的方向控制为设计氢化催化剂提供了新的策略。
引言
近年来的研究对Cu基催化剂活性位点的揭示表明Cu0和Cu+之间的协同作用是优化催化性能的主导因素,平衡Cu+/Cu0比率是提高铜基催化剂催化活性的常见策略,但仍然受到低原子利用率和电荷分布固有性质的限制。因此,本工作采用液相氢供体(甲酸、水合肼),通过原位还原方法成功制备了空间分离的板状Cu+簇和贵金属位点的负载型催化剂。例如,甲酸(FA)在板状Cu0团簇上分解形成活性氢物种(FA在催化剂表面分解),然后通过Cu金属位点周围的活性氢物种还原Pd4+物种,同时还原Cu2+到Cu+物种。Pd物种预计将锚定在Cu金属位点附近的区域,从而实现氧化物载体上Pd和Cu金属位点等密度的趋势变化。结果,Cu100Pd1/CeO2-FA(Pd/Cu=1:100,mol/mol)对HMF选择性加氢表现出优异的催化活性。
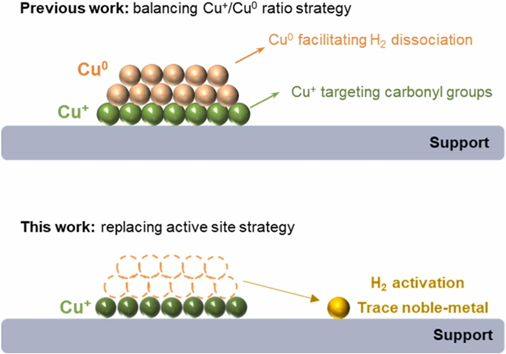
Scheme 1. Schematic illustration of replacing Cu0 active site.
图文导读
催化剂合成过程中,首先采用浸渍法制备CuO/CeO2,在此基础上通过原位还原法制备Cu100Pd1/CeO2-FA和Cu100Pd1/CeO2-NH。结合AC-HAADF-STEM和EDS元素谱图,Cu金属主要以板状Cu+团簇形式存在于氧化铈载体表面,而传统通过共浸渍-氢气还原方法制备的Cu100Pd1/CeO2-H(500)催化剂上的铜金属则以含有Cu0−Cu+界面的Cu纳米团簇形式存在。此外,钯金属以单原子锚定在Cu100Pd1/CeO2-FA上,但在Cu100Pd1/CeO2-H(500)上则以纳米团簇的形式出现,这一点得到了DRIFT-CO光谱的有力证实。Cu100Pd1/CeO2-FA和Cu/CeO2-FA的XANES光谱以及原位XPS(Cu L3VV俄歇谱)显示Cu100Pd1/CeO2-FA和Cu/CeO2-FA加氢反应过程中,板状Cu团簇均保持+1价态。EXAFS表征揭示了催化剂中Pd−O键的主导性以及Pd−Pd键的缺失,从而坚实的证实了Pd物种在Cu100Pd1/CeO2-FA催化剂上原子分散。
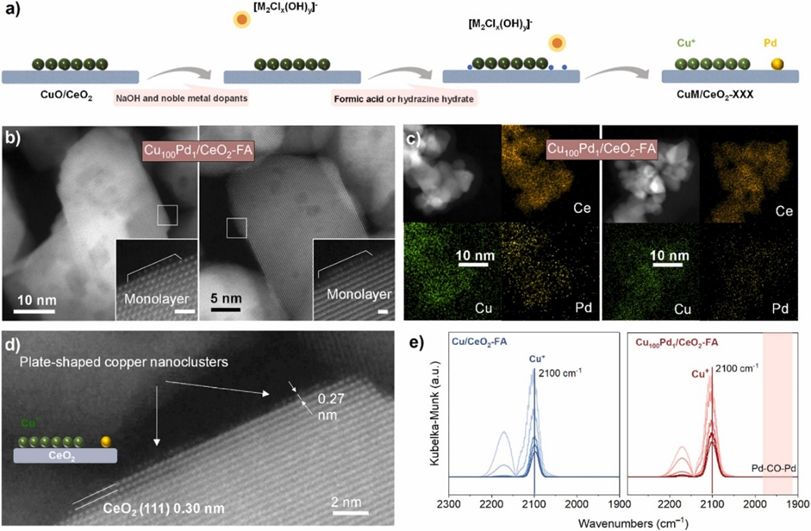
图1. (a) Cu100Pd1/CeO2-FA合成示意图。(b,c) Cu100Pd1/CeO2-FA的像差校正HAADF-STEM图像和EDS元素映射图像。(d) Cu100Pd1/CeO2-FA上铜簇的原子分辨HAADF-STEM图像。(e) Cu/CeO2-FA和Cu100Pd1/CeO2FA在2300−1900cm−1范围内流动N2的原位CO-DRIFTS谱。
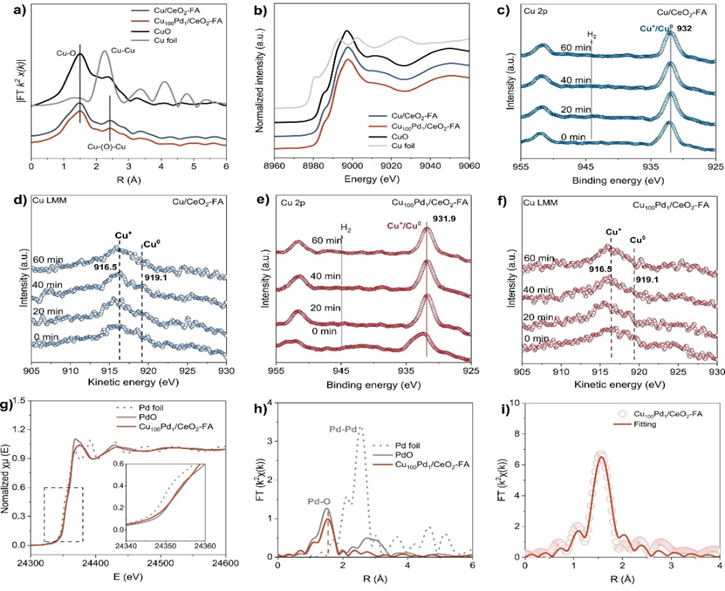
图2. (a) 不同催化剂和参考样品在Cu K边缘的XANES曲线。(b) R空间中不同催化剂的EXAFS光谱和Cu K边缘的参考。(c,e) Cu/CeO2-FA和Cu100Pd1/CeO2-FA的原位Cu 2p XPS谱。(d,f) Cu/CeO2-FA和Cu100Pd1/CeO2-FA的原位Cu LMM XPS光谱。(g) 归一化Pd K边XANES光谱。(h) k3加权Pd K边缘EXAFS光谱的傅里叶变换。(i) 合成的Cu100Pd1/CeO2-FA催化剂的EXAFS最佳拟合结果。
催化性能
图3. (a)表明,Cu金属和微量Pd金属都是完成高催化活性所必需的。反应动力学实验证明Cu100Pd1/CeO2-FA催化剂的表观活化能(54kJ/mol)明显低于Cu/CeO2-FA催化剂的表观活化能(77kJ/mol),表明痕量钯能显著降低表观活化能。在温和的反应条件下,Cu100Pd1/CeO2-FA表现出非常高的生产率,甚至超过了部分贵金属催化剂。本研究还通过改变氢供体(甲酸、水合肼)、贵金属(Pd、Pt和Ru)和载体(CeO2、In2O3和ZnO)合成了一系列铜基催化剂,所有含有痕量贵金属的铜基催化剂都表现出增强的C=O键的加氢活性和选择性。
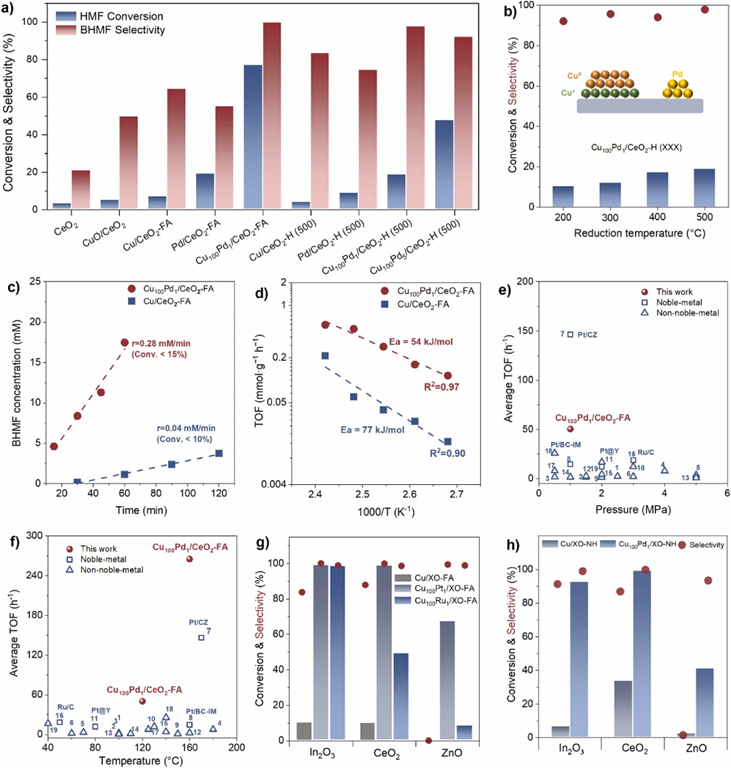
图3. (a) 催化剂的催化性能。(b) 不同还原温度制备的Cu100Pd1/CeO2-H催化剂的催化评价。反应条件:乙醇 20mL、HMF 0.3g、催化剂 0.1g、120℃、1MPa H2、0.5h。(c) BHMF相对于Cu100Pd1/CeO2-FA和Cu/CeO2-FA的形成速率。反应条件:乙醇 20mL、HMF 0.3g、催化剂 0.02g、120℃、1MPa H2。(d) HMF氢化过程中Cu100Pd1/CeO2-FA和Cu/CeO2-FA的阿伦尼乌斯图。(e)和(f) Cu100Pd1/CeO2-FA与近五年报道的催化剂在HMF加氢中的催化活性比较。完整数据如表S2所示。(1) Cu/SiO2; (2) Cu-ZnO; (3) Ni1.5Co1; (4) 10Ni/SiO2; (5) meso-Cu/Al2O3; (6) Ni/HSAG; (7) Pt/CZ; (8) Pt/BC-IM; (9) Cu@C-POP; (10) 20 %Cu-Al2O3; (11) Pt@Y; (12) 10 %-Ni/HC; (13) Raney Co; (14) Co@C; (15) CuNPs@ZIF-8; (16) Ru/C; (17) CuHTO; (17) Cu/SiO2; (18) Co/Al2O3。(g) 通过原位还原法制备的不同贵金属(Pt和Ru)的催化剂的催化性能。(h)水合肼还原制备的催化剂的催化性能。反应条件:乙醇 20mL,HMF 0.3g,催化剂 0.1g,120℃,1MPa H2,1h。
催化机理和氢迁移
为了从进一步了解板状Cu+团簇和Pd位点的催化功能,我们首先通过原位红外光谱揭示HMF分子分别在Cu+团簇和Pd位点上的吸附构型。密度泛函理论计算证明Cu100Pd1/CeO2-FA催化剂上的Cu位点和Pd位点分别是HMF垂直吸附和H2吸附的活性位点。也就是说明,单原子Pd位点是氢气活化中心,而板状Cu+团簇是活性氢添加到不饱和C=O键上的活性位点。结合H/D同位素动力学实验,HMF在Cu100/CeO2-FA催化剂上的加氢速率决定步骤是H2的活化和解离,而在Cu100Pd1/CeO2-FA催化剂上速率决定步骤改为表面加氢过程。
使用WO3作为显色剂来证明氢溢出的存在,H2在Pd原子上解离吸附产生的H+和H-会自发迁移到不容易活化H2分子的板状Cu+团簇上。另外,原位XPS展示Cu100Pd1/CeO2-FA表明Ce3+物种的形成速度更快,其结合能位移更大,这进一步证明痕量Pd单原子加速了H2分子的活化和解离。
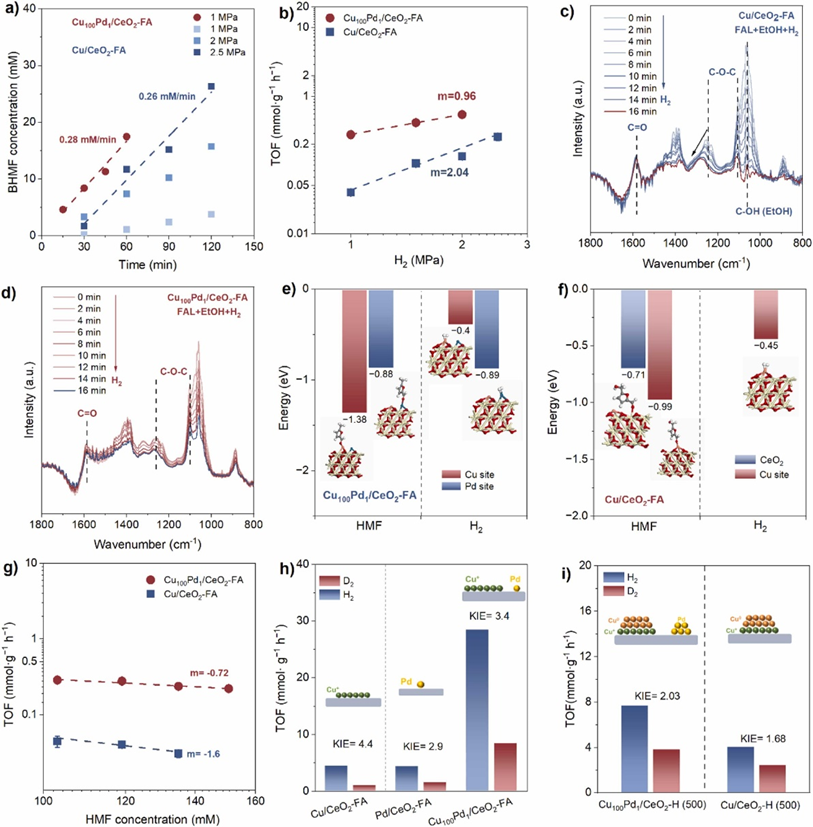
图4. (a) 在不同的H2压力下Cu100Pd1/CeO2-FA和Cu/CeO2-FA上的BHMF形成速率。(b) HMF中C=O键氢化的H2压力反应顺序。(c,d) 原位FT-IR光谱记录了Cu/CeO2-FA(c)和Cu100Pd1/CeO2-FA(d)在H2/Ar流中每隔一段时间的糠醛吸附模式。(e,f) 不同吸附构型下Cu/CeO2-FA和Cu100Pd1/CeO2-FA的H2和HMF吸附能。(g)HMF氢化的HMF浓度的反应顺序。(h) H2和D2中Cu/CeO2-FA、Pd/CeO2-FA和Cu100Pd1/CeO2-FA的BHMF形成速率的TOF。(i) H2和D2中Cu/CeO2-H(500)和Cu100Pd1/CeO2-H(500)的BHMF形成速率的TOF。
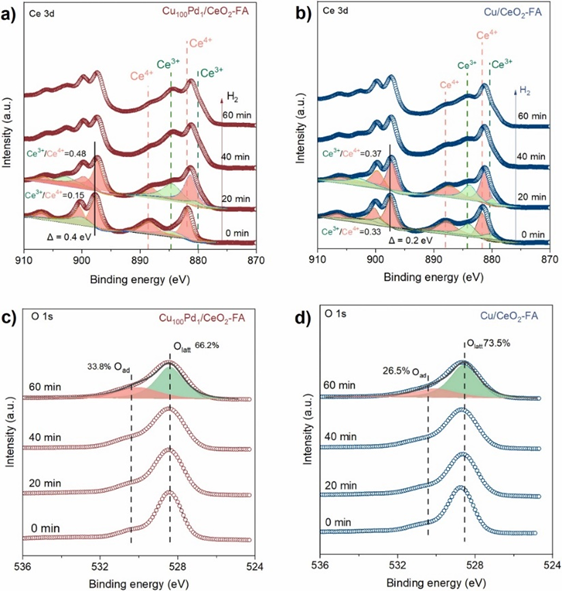
图5. (a,b) Cu100Pd1/CeO2-FA和 Cu/CeO2-FA的原位Ce 3d XPS谱。(c,d) Cu100Pd1/CeO2-FA和Cu/CeO2-FA的原位O 1s XPS谱。